
Como saben los fieles seguidores de este blog, en SCIENTIA le he dedicado muchos posts a las enfermedades raras. Creo que es importantísimo divulgar no solo en qué consisten estas enfermedades sino también los nuevos avances científicos que se producen en el campo de estas patologías. Por ello es posible que a muchos les extrañase que ayer, Día Mundial de las Enfermedades Raras, no publicase nada al respecto y sí lo haga hoy. El motivo está claro. Valoro muchísimo jornadas como las de ayer donde todo el mundo informó sobres estas patologías pero no me gusta que sobre este tema se escriba, se hagan galas o se hable en televisión solamente cada 365 días …y hasta el año siguiente.

Dentro de las enfermedades raras hay un grupo que me llama poderosamente la atención y al que dedico parte de mis labores investigadoras en la Universidad de Murcia: las enfermedades por almacenamiento de lípidos. Este tipo de patologías son un grupo de trastornos metabólicos heredados que se caracterizan por la acumulación de altas concentraciones de lípidos en algunas células y tejidos del cuerpo. Esta acumulación puede provocar graves daños tisulares y celulares, particularmente en el cerebro, el sistema nervioso periférico, el hígado, el bazo y la médula ósea.
En Scientia ya hemos hablado de algunas enfermedades raras que forman parte de este grupo. Me refiero a la enfermedad de Gaucher (causada por una deficiencia de la enzima glucocerebrosidasa) o la enfermedad de Niemann-Pick (causada por mutaciones en los genes NPC1 y NPC2 que codifican proteínas implicadas en la normal regulación del tráfico lipídico intracelular). En el post de hoy quiero hablarles de la enfermedad de Wolman, también conocida como deficiencia de la lipasa ácida, y que fue descrita en 1954 por el médico Moshe Wolman. Esta enfermedad afecta a menos de 1 de cada 100,000 nacimientos.
Existe una importante heterogeneidad clínica entre los pacientes con este déficit enzimático que va desde la forma más grave, en que la mayoría de los pacientes fallecen durante el primer año de vida (enfermedad de Wolman), a una forma relativamente benigna de enfermedad de depósito de ésteres de colesterol (CESD), la cual ha sido identificada en la edad adulta. Me centraré en la forma más grave.
Enzima lipasa ácida humana
La reducción en más de un 90% de la actividad de la enzima lipasa ácida, una glicoproteína encargada de la hidrólisis de los ésteres de colesterol y triglicéridos en el paso de sangre periférica a la célula, conduce a un almacenamiento masivo de triglicéridos y ésteres de colesterol principalmente el cerebro, hígado y bazo. La deficiencia enzimática se produce a causa de graves mutaciones del gen de la lipasa ácida (LIPA o LAL), localizado en el cromosoma 10 y que porta la información genética codificada para generar esta proteína.
A pesar de que al nacer los niños que padecen esta enfermedad presentan un aspecto normal, no suelen alcanzar el año de vida. Rápidamente manifiestan deterioro de capacidades mentales, agrandamiento del hígado (hepatomegalia) y del bazo (esplenomegalia). También aparecen alteraciones intestinales como esteatorrea (eliminación de grasas por las heces) y distensión del abdomen. Otros síntomas comunes son anemia y depósitos de calcio en las glándulas suprarrenales, provocando que se endurezcan.
A día de hoy no existe una forma efectiva de combatir esta patología que afecta a un muy reducido grupo de población. De hecho, la mayoría de los tratamientos van dirigidos a paliar los síntomas que produce o a abordarla indirectamente. Un ejemplo es la recomendación de darle a los niños que sufran la enfermedad de Wolman fórmulas que no contengan triglicéridos y ésteres de colesterol a la misma vez que se les administra una nutrición suficiente vía parenteral que incluya todas las vitaminas incluyendo las liposolubles. Otro ejemplo de tratamiento son los trasplantes de hígado o médula ósea.
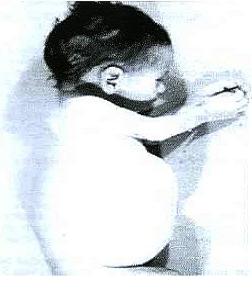
Niño con la enfermad de Wolman. Fuente: Ecured.
¿Y por qué he elegido hoy la enfermedad de Wolman para reivindicar la investigación en enfermedades raras? Por una reciente decisión en EEUU de la “Food and Drug Administration (FDA)” que me ha gustado mucho y que permite abordar la enfermedad desde otro punto de vista. Como les he comentado los tratamientos actuales van más encaminados a paliar los síntomas de la enfermedad que a curar la misma. En este caso la entidad norteamericana ha aprobado el uso de pollos modificados genéticamente (sí, pollos transgénicos) para enfrentarse a la enfermedad de Wolman de una forma directa.
La modificación genética de los pollos ayudará a que produzcan huevos en cuya clara aparezcan grandes cantidades de la enzima lipasa ácida. Una vez que los pollos han “puesto” los huevos se extrae la clara y se purifica la enzima. Luego “solo” queda usar la enzima para desarrollar un fármaco con el que tratar a los niños a los que se les haya detectado el Síndrome de Wolman. Con ello se logrará suplir sus carencias en la enzima lipasa ácida enzima y evitar la acumulación de lípidos. Hay que recordar que estos fármacos destinados a combatir las enfermedades raras reciben el nombre de ‘medicamentos huérfanos’ ya que carecen de interés para la industria farmacéutica tradicional.
En el caso que hoy nos ocupa, el fármaco en cuestión recibe el nombre de Kanuma y se administra vía venosa. Concretamente Kanuma es el nombre comercial de una enzima, sebelipasa alfa, producida en la clara del huevo de pollos modificados genéticamente. Su uso es un ejemplo de lo que se conoce como Terapia de Reemplazo Enzimático (suministrar al paciente la proteína éxogena que en su organismo está siendo sintetizada de forma anormal) y la empresa responsable de la comercialización es Alexion. Estudios clínicos realizados con niños que sufren la enfermedad de Wolman y a los que se les administró el fármaco Kanuma han dado lugar a resultados satisfactorios.

¿Y se pueden comer esos pollos? No. La aprobación de la FDA está referida al uso de los pollos como “medicamentos animales” y no como alimentos propiamente dichos. Estrategias similares se han empleado para modificar genéticamente cabras y conejos que producen, respectivamente, proteínas recombinantes que se emplean para prevenir trombos (fármaco ATryn) o para tratar ataques agudos de angioedema hereditario de tipo I (fármaco Ruconest).
Estimados lectores, la investigación en enfermedades raras es la clave para intentar solucionar la mayoría de los problemas de las personas que sufren este tipo de patologías. Pero no nos equivoquemos. Sin el apoyo de estamentos públicos y privados, de la industria farmacéutica y, sobre todo, sin la sensibilidad de todos , las enfermedades raras seguirán siendo un drama tanto para pacientes, familiares y cuidadores. Es cierto que existen otras patologías cuya prevalencia e incluso índice de mortalidad son mucho mayores por lo que se destinan más ayudas a su estudio, pero también es cierto que los afectados por las enfermedades raras, a pesar de estar en franca minoría, son igualmente personas que sufren terribles enfermedades, con un rechazo social tremendo y son merecedores de, al menos, la misma atención que otros enfermos.
Pero si investigar es prioritario, no lo es menos divulgar los resultados obtenidos y la importancia de los mismos. Eso sí, no nos limitemos a hacerlo cada 28 de febrero…
Jose
Nota: Este post forma parte de la serie “Proteínas del siglo XXI”
– Post sobre enfermedades Raras publicados en Scientia:
- El sorprendente caso de la enfermedad rara que te inmuniza ante el virus del Ébola
- El resveratrol y el síndrome de Hutchinson-Gilford (progeria): una puerta abierta a la esperanza
- Raras pero no invisibles…algo más que un post
- Encapsulando a Niemann-Pick
- ¿Hay que investigar en las “enfermedades raras”?
- Las zanahorias transgénicas, Gaucher y el nuevo gobierno francés
- Raras pero no invisibles…un proyecto hecho realidad
– Conferencias sobre enfermedades Raras publicados en Scientia:



Gracias….no hay cosa más grande para un padre ver que alguien utiliza sus medios para difundir los problemas que tienen nuestros pequeños con enfermedades raras (o poco frecuentes, poco conocidas o infradiagnosticadas) en nuestro caso, Dravet, mutación genetica SCN1A.
Hola JM,
sólo comentar que los medicamentos huerfanos no es que carezcan de interés para la industria farmacéutica, es que los costes de la investigación no pueden ser recuperados vendiendo las medicinas en farmacias. Deberían salir a un precio muy elevado ya que hay muy pocos pacientes.
Lo que sucede es que cuando una farmaceutica descubre en una fase temprana de investigación que una nueva molécula puede ser efectiva en una enfermedad rara, el laboratorio contacta las autoridades (EMEA, FDA) para llegar a un acuerdo de «medicamento huerfano» y seran los paises directamente quienes se haran cargo de la factura.
Tras el acuerdo, se continua con las siguientes fases de la investigación.
Simplemente gracias.
Pingback: Proteínas del siglo XXI | SCIENTIA
Pingback: Pollos transgénicos y la enfermedad rara de Wolman
Pingback: Pollos transgénicos y la enfermedad rara...
Pingback: Pollos transgénicos y la enfermedad rara...